File Formats
This page describes the fileformats as used in the Illumina genome
analyzer.
Intensity files
These are the files that can be found in the directory
s_#lane_#tile_nse.txt
These files are generated during the cluster detection and are tab seperated.
The content of the file is not well documented but it seems to cover the variance within each cluster.
cycle 1 cycle 2 cycle 3
lane tile x y a c g t a c g t a c g t ...
2 47 219 303 9.9 9.9 4.9 4.6 15.0 16.9 6.0 8.8 14.9 17.7 7.8 7.8 ...
s_#lane_#tile_int.txt
The intensity files contain the average intensity for each cluster on each of the 4 images. Below is an example. These
intensities are non-normalized. So crosstalk is still present.
cycle 1 cycle 2 cycle 3
lane tile x y a c g t a c g t a c g t ...
2 47 219 303 64.8 69.4 1567.9 525.7 790.7 597.9 45.8 32.2 178.1 630.8 46.1 29.6
Base called files
s_#lane_#tile_sig2.txt
These are the files afte crosstalk correction has been performed. If interested in the intensities this is the file that should be used. Its format is again tab seperated. One row per cluster.
cycle 1 cycle 2 cycle 3
lane tile x y a c g t a c g t a c g t ...
2 47 219 303 -6.5 3.5 1056.5 -24.7 661.4 12.7 17.1 13.9 -23.2 679.4 17.6 10.2
s_#lane_#tile_seq.txt
The sequence file that will list for each cluster the bases that have been called. If a base could not be called a '.' is used.
lane tile x y sequence
2 47 219 303 GACATTATGGGTCTGCAAGCTGCTTATGCTAATTTG
2 47 223 1924 GGTGTGGTTGATATTTTTCATGGTATTGATAAAGCT
2 47 621 348 GGAAGTAGCGACAGCTTGGTTTTTAGTGAGTTGTTC
2 47 892 162 GCTTCCATAAGCAGATGGATAACCGCATCAAGCTCT
2 47 1473 657 GCTTTATCAAGATAATTTTTCGACTCATCAGAAATA
2 47 670 345 GTCAATCCTGACGGTTATTTCCTAGACAAATTAGAG
2 47 1787 755 G...................................
s_#lane_#tile_prb.txt
The probability file contains for each cluster the probabilities that
the specific bases were called properly ? The file format is explained
in detail
here.
Each line corresponds to the cluster found at the same line in the _seq, _sig2, _int and _qhg files.
The tabs are at unexpected places: between each cycle and not between each number. The numbers are simply seperated by spaces.
cycle 1 cycle 2 cycle 3 cycle 4
a c g t a c g t a c g t a c g t ...
-40 -40 40 -40 40 -40 -40 -40 -40 40 -40 -40 40 -40 -40 -40 ...
s_#lane_#tile_qhg.txt
The quality metrics that can afterwards be used for filtering the data
Lane; tile; xPos; yPos; chastity, purity;
similarity; neighbour; neighbourhood
Lane Tile x y chastity purity similarity neighbour neighbourhood
2 47 219 303 0.67 0.82 -0.43 3.00 70696:30892:61591:23697:37453:32059:85214:54989
Eland aligned files
s_#lane_#tile_all.png
This is a file that reports the average intensity over each of the
called bases for the specified tile and lane.
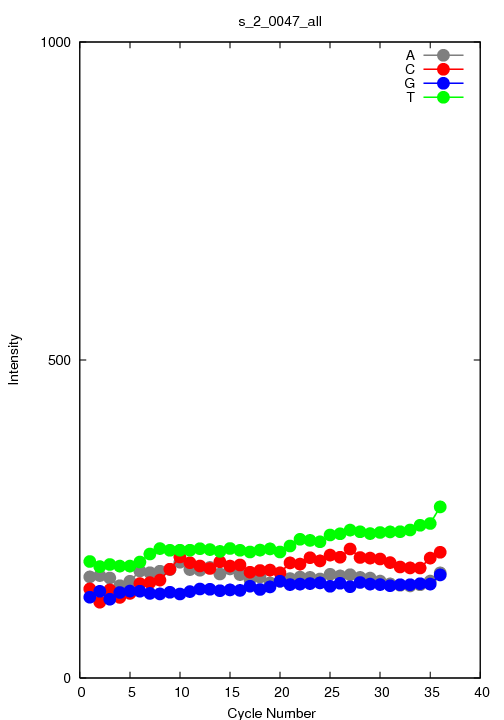
s_#lane_#tile_all.txt
Contains the statistics that are used to generate the various plots.
# Clusters: Filtered 63791 Original 100455
#
#Lane Cycle All A All C All G All T Call A Call C Call G Call T Num A Num C Num G Num T Num X
2 1 158.5 141.9 127.0 183.2 575.7 546.2 595.1 585.7 17055 14516 13525 18695 0
2 2 160.5 120.8 136.4 174.8 546.8 525.8 575.1 550.7 17442 12941 14551 18857 0
2 3 156.6 140.7 123.8 178.3 549.2 534.6 558.3 548.7 17000 14412 13514 18865 0
2 4 144.6 128.2 134.5 175.6 516.9 500.6 553.2 535.4 16491 14249 14553 18498 0
2 5 151.2 134.7 137.0 175.8 518.1 522.6 550.4 528.2 17284 12970 14755 18782 0
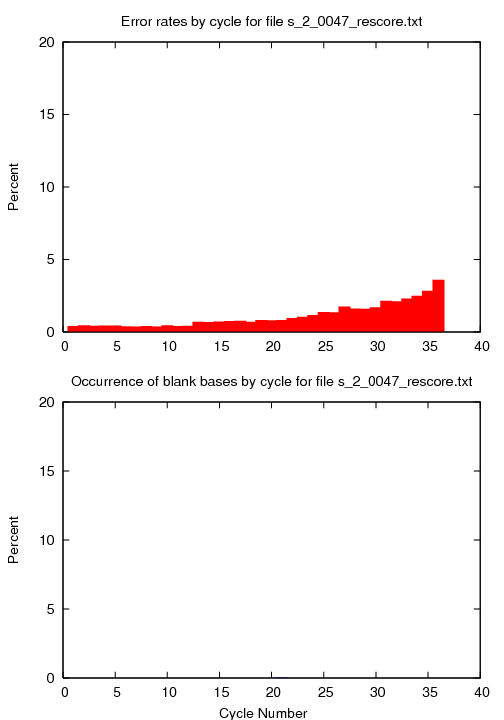
s_#lane_#tile_errors.png
This image reports the number of sequences that could be directyle/one mismatch/two mismatches be aligned after x cycles. Below is an example.
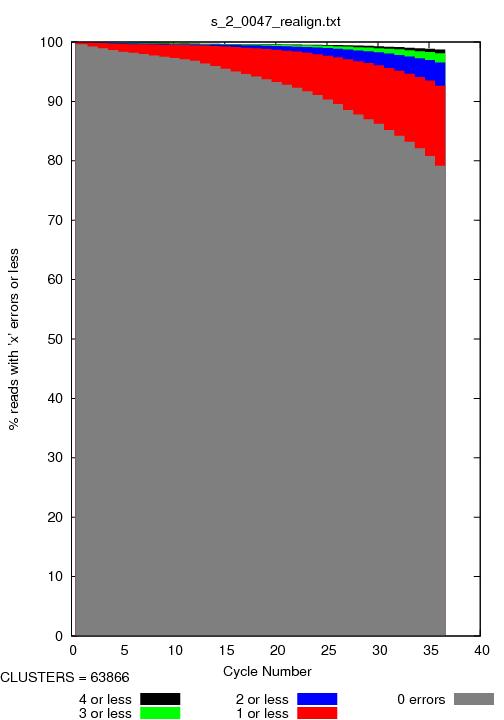
s_#lane_#tile_rescore.png
This image reports the percentage of errors per position in the alginment.
Below is an example.
s_#lane_#tile_rescore.txt
This is actually an interesting file that reports on the filtering of the data. It contains multiple sections. Below is a demonstration of such a file shortened somewhat by removing cycles 2-35.
#RUN_TIME Thu Jun 26 20:47:35 2008
#SOFTWARE_VERSION @(#) $Id: score.pl,v 1.2 2008-05-30 07:36:06 wernersa Exp $
# Lane 2 : Tile 47 : Quality Filtered
2233116 bases of sequence found
16279 were errors
20 were blanks
0.73 percent error rate
0.00 percent blank rate
unique alignments : 62031 (total score 3937852)
cycles : 36
Breakdown of errors by cycle
Cycle: Err pc: Blank pc:
1 0.19 0.00
36 3.14 0.00
Error rate relative to reference base (including blanks)
(Given a reference base, what is it sequenced as?)
Really: Read as:
N pc A pc C pc G pc T pc
A 0.00 99.46 0.28 0.12 0.14
C 0.00 0.21 99.41 0.03 0.35
G 0.00 0.24 0.13 98.37 1.26
T 0.00 0.04 0.15 0.10 99.70
Error rate relative to reference base (excluding blanks)
(Given a reference base, what is it sequenced as?)
Really: Read as:
A pc C pc G pc T pc
A 99.46 0.28 0.12 0.14
C 0.21 99.41 0.03 0.35
G 0.24 0.13 98.37 1.26
T 0.04 0.15 0.10 99.70
Error rate relative to sequenced base
(Given a sequenced base, what was it really?)
Read as: Really:
A pc C pc G pc T pc
N 30.00 25.00 10.00 35.00
A 99.57 0.18 0.21 0.05
C 0.33 99.33 0.13 0.20
G 0.14 0.03 99.69 0.14
T 0.13 0.26 0.95 98.66
Breakdown of errors by nucleotide
Read As: Really:
A pc C pc G pc T pc
N 6 5 2 7
A 581763 1042 1200 266
C 1650 494665 656 1011
G 683 165 489812 680
T 844 1744 6258 650577
Full breakdown of errors by cycle and nucleotide
Cycle: Read As: Really:
A ct C ct G ct T ct
1 N 0 0 0 0
1 A 16515 18 23 0
1 C 4 14118 3 17
1 G 12 1 13100 10
1 T 4 18 5 18183
36 N 0 0 0 0
36 A 15764 51 41 25
36 C 171 13740 90 46
36 G 43 17 12627 40
36 T 125 142 1156 17922
Error rate relative to reference by cycle/nucleotide
Cycle: Read As: Really:
A ct C ct G ct T ct
@1 N 0.000 0.000 0.000 0.000
@1 A 0.999 0.001 0.002 0.000
@1 C 0.000 0.997 0.000 0.001
@1 G 0.001 0.000 0.998 0.001
@1 T 0.000 0.001 0.000 0.999
@36 N 0.000 0.000 0.000 0.000
@36 A 0.979 0.004 0.003 0.001
@36 C 0.011 0.985 0.006 0.003
@36 G 0.003 0.001 0.908 0.002
@36 T 0.008 0.010 0.083 0.994
Information Content By Cycle
Bases this cycle Bases so far
Cycle: Equiv info: Align: Total: Equiv info: Align: Total:
~1 61363.11 62031 63791 61363.11 62031 63791
~36 55349.21 62031 63791 2161274.97 2233096 2296454
The 20 most common words with 2 blanks or less:
Ranking Occurrences Words
1 21 GTTTGATGAATGCAATGCGACAGGCTCATGCTGATG TCTCATATTGGCGCTACTGCAAAGGATATTTCTAAT
2 20 CTTGCTATTGACTCTACTGTAGACATTTTTACTTTT
3 18 ACTGATGCTGCTTCTGGTGTGGTTGATATTTTTCAT AGAACGTTTTTTACCTTTAGACATTACATCACTCCT AGATGGATAACCGCATCAAGCTCTTGGAAGAGATTC
4 17 CGATTAGAGGCGTTTTATGATAATCCCAATGCTTTG
5 16 9 sequences
6 15 24 sequences
7 14 28 sequences
8 13 56 sequences
9 12 104 sequences
10 11 161 sequences
11 10 306 sequences
12 9 447 sequences
13 8 610 sequences
14 7 896 sequences
15 6 1246 sequences
16 5 1529 sequences
17 4 1677 sequences
18 3 1676 sequences
19 2 1730 sequences
20 1 10463 sequences
The 3 most common blank patterns (N=any nonblank character)
Ranking Occurrences Words
1 63769 NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
2 20 NNNNNNNNNNNNNNNNNNNN.NNNNNNNNNNNNNNN
3 1 NNNNNNNNNNNNNNNNNNNNNNNN.NNNNNNNNNNN NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN.NNN
Log likelihood scores
Cycle: Read As: Really:
A C G T
>1 N -47 -47 -47 -47
>1 A 260 -296 -285 -47
>1 C -354 276 -367 -291
>1 G -303 -411 275 -311
>1 T -365 -300 -356 282
>36 N -47 -47 -47 -47
>36 A 212 -249 -258 -280
>36 C -190 165 -219 -248
>36 G -246 -287 210 -250
>36 T -218 -213 -119 110
Cumulative errors by cycle
Cycle: 1 2 3 4 5
!1 61916 62031 62031 62031 62031
!36 50087 58673 61221 61866 62010
s_#lane_#tile_score.txt
Various statistics of this tile and lane before quality filtering.
#RUN_TIME Thu Jun 26 20:47:35 2008
#SOFTWARE_VERSION @(#) $Id: score.pl,v 1.2 2008-05-30 07:36:06 wernersa Exp $
# Lane 2 : Tile 47 : Raw
2691216 bases of sequence found
33142 were errors
134 were blanks
1.23 percent error rate
0.00 percent blank rate
unique alignments : 74756 (total score 4168089)
cycles : 36
Breakdown of errors by cycle
Cycle: Err pc: Blank pc:
1 0.50 0.10
36 4.25 0.00
Error rate relative to reference base (including blanks)
(Given a reference base, what is it sequenced as?)
Really: Read as:
N pc A pc C pc G pc T pc
A 0.01 98.95 0.50 0.20 0.35
C 0.01 0.44 98.96 0.11 0.47
G 0.00 0.32 0.34 97.74 1.60
T 0.01 0.14 0.34 0.29 99.23
Error rate relative to reference base (excluding blanks)
(Given a reference base, what is it sequenced as?)
Really: Read as:
A pc C pc G pc T pc
A 98.96 0.50 0.20 0.35
C 0.44 98.97 0.11 0.47
G 0.32 0.34 97.74 1.60
T 0.14 0.34 0.29 99.24
Error rate relative to sequenced base
(Given a sequenced base, what was it really?)
Read as: Really:
A pc C pc G pc T pc
N 26.87 27.61 10.45 35.07
A 99.20 0.38 0.27 0.15
C 0.58 98.63 0.34 0.45
G 0.23 0.11 99.27 0.38
T 0.31 0.36 1.20 98.13
Breakdown of errors by nucleotide
Read As: Really:
A pc C pc G pc T pc
N 36 37 14 47
A 697767 2655 1931 1076
C 3502 592725 2048 2675
G 1377 669 586074 2245
T 2461 2838 9576 781374
Full breakdown of errors by cycle and nucleotide
Cycle: Read As: Really:
A ct C ct G ct T ct
1 N 22 19 8 25
1 A 19825 88 40 13
1 C 14 16733 12 36
1 G 21 11 15886 61
1 T 24 41 10 21867
36 N 0 0 0 0
36 A 18785 109 77 53
36 C 284 16441 184 113
36 G 84 53 14815 78
36 T 315 245 1578 21507
Error rate relative to reference by cycle/nucleotide
Cycle: Read As: Really:
A ct C ct G ct T ct
@1 N 0.001 0.001 0.001 0.001
@1 A 0.996 0.005 0.003 0.001
@1 C 0.001 0.991 0.001 0.002
@1 G 0.001 0.001 0.996 0.003
@1 T 0.001 0.002 0.001 0.994
@36 N 0.000 0.000 0.000 0.000
@36 A 0.965 0.006 0.005 0.002
@36 C 0.015 0.976 0.011 0.005
@36 G 0.004 0.003 0.890 0.004
@36 T 0.016 0.015 0.095 0.989
Information Content By Cycle
Bases this cycle Bases so far
Cycle: Equiv info: Align: Total: Equiv info: Align: Total:
~1 72766.19 74682 100268 72766.19 74682 100268
~36 64041.84 74756 98558 2548406.67 2691082 3537981
The 22 most common words with 2 blanks or less:
Ranking Occurrences Words
1 22 TCTCATATTGGCGCTACTGCAAAGGATATTTCTAAT
2 21 GTTTGATGAATGCAATGCGACAGGCTCATGCTGATG
3 20 CTTGCTATTGACTCTACTGTAGACATTTTTACTTTT
4 19 AGAACGTTTTTTACCTTTAGACATTACATCACTCCT TGCTTTTGATGCCGACCCTAAATTTTTTGCCTGTTT
5 18 ACTGATGCTGCTTCTGGTGTGGTTGATATTTTTCAT AGATGGATAACCGCATCAAGCTCTTGGAAGAGATTC CGATTAGAGGCGTTTTATGATAATCCCAATGCTTTG GTATTCTGGCGTGAAGTCGCCGACTGAATGCCAGCA TGACTATTGACGTCCTTCCTCGTACGCCGGGCAATA
6 17 9 sequences
7 16 14 sequences
8 15 24 sequences
9 14 50 sequences
10 13 102 sequences
11 12 124 sequences
12 11 228 sequences
13 10 365 sequences
14 9 534 sequences
15 8 720 sequences
16 7 1002 sequences
17 6 1328 sequences
18 5 1531 sequences
19 4 1638 sequences
20 3 1502 sequences
21 2 1778 sequences
22 1 39095 sequences
The 30 most common blank patterns (N=any nonblank character)
Ranking Occurrences Words
1 97289 NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
2 1661 N...................................
3 173 N....NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
4 134 N...NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
5 128 .NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
6 79 N.....NNNNNNNNNN....NNNNNNNNNNNNNNNN
7 67 NN..................................
8 60 N.....NNNNNNNNNNN..NNNNNNNNNNNNNNNNN
9 58 ....................................
10 49 N.....NNNNNNNNNN...NNNNNNNNNNNNNNNNN
11 46 N.......................NNNN......NN
12 43 N.......NNNNN.......NNNNNNNNNNN..NNN
13 36 N.....NNNNNNNNN.....NNNNNNNNNNNNNNNN
14 34 N........NNNN.........NNNNNNNN...NNN
15 30 N.........NN...........NNNNNN.....NN
16 27 N.......................NNNN......N.
17 26 NNNNNNNNNNNNNNNNNNNN.NNNNNNNNNNNNNNN
18 22 N..........N...........NNNNNN.....NN N.......NNNNN........NNNNNNNNN...NNN N.......NNNNN.......NNNNNNNNNNN.NNNN
19 19 N......NNNNNN.......NNNNNNNNNNN.NNNN NN.........................N........
20 18 N.....NNNNNNNNNNNN.NNNNNNNNNNNNNNNNN
21 17 N......NNNNNNNN.....NNNNNNNNNNNNNNNN
22 16 N........NNN...........NNNNNN....NNN N.......NNNNN.......NNNNNNNNNN...NNN NN.NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
23 15 N.........................N.........
24 13 N........................NN......... N......................NNNNNN.....NN N.......NNNNN.........NNNNNNNN...NNN
25 12 N.......................NNNNN.....NN
26 11 N......NNNNNN.N.....NNNNNNNNNNN.NNNN N.....NNNNNNNNNNNNNNNNNNNNNNNNNNNNNN NN.....NNNNNNNN.......NNNNNNNNNNNNNN
27 10 N........................NNN........ NNN..NNNNNNNNNN..N...NNNNNNNNNNNNNNN
28 9 N........................NNN......N. N........NNN..........NNNNNNN....NNN N..NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN NNN.NNNNNNNNNNNNNNNN.NNNNNNNNNNNNNNN
29 5 N......NNNNNNNN.....NNNNNNNNNNN.NNNN NN........................NN........ NN.....NNNNNNNN.......NNNNNNNNN.NNNN NNN..NNNNNNNNNNNNN...NNNNNNNNNNNNNNN
30 4 NN.....NNNNNN..........NNNNNNNN.NNNN
Log likelihood scores
Cycle: Read As: Really:
A C G T
>1 N -37 -46 -91 -29
>1 A 214 -235 -269 -318
>1 C -307 243 -314 -266
>1 G -288 -316 223 -241
>1 T -296 -272 -334 246
>36 N -47 -47 -47 -47
>36 A 189 -223 -239 -255
>36 C -177 145 -196 -217
>36 G -225 -245 183 -228
>36 T -186 -198 -114 100
Cumulative errors by cycle
Cycle: 1 2 3 4 5
!1 74311 74756 74756 74756 74756
!36 54261 66703 72317 74193 74601
PhageAlign Output
The s_N_TTT_align.txt files contain the unfiltered first-pass
alignments for a give tile. The s_N_TTTT_prealign.txt contains a
recalibration of the aligned sequences, thereby taking into account
the errormodel created based on the alignments (_align.txt and
_score.txt). The file s_N_realign.txt consists of alignments in
s_N_TTT_prealign.txt that do pass the filter criteria.
Lane summaries
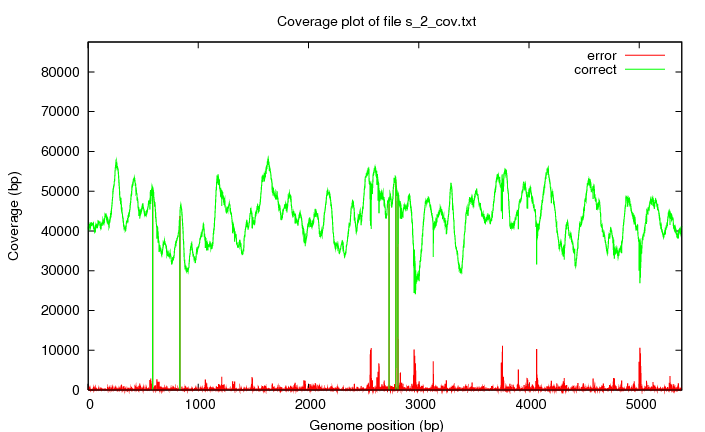
s_#lane_cov.png
The coverage of the bases compared to the genome.
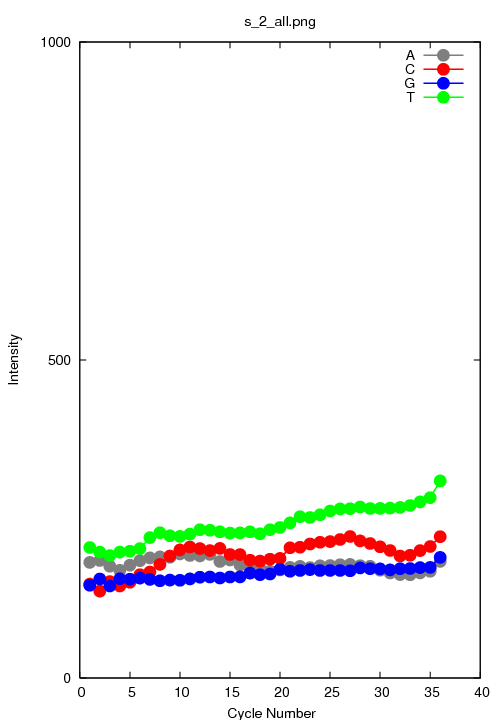
s_2_all.png
The average intensity over all tiles.
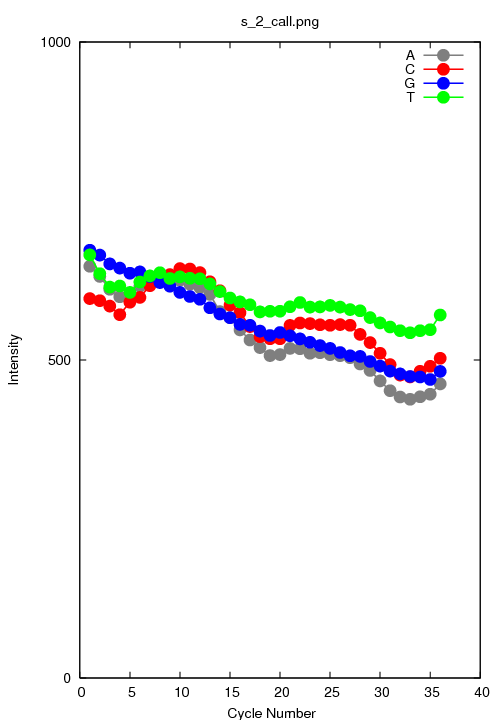
s_2_call.png
The average intensity of only the called bases.
s_2_calsaf.txt
s_2_eland_extended.txt
s_2_eland_multi.txt
s_2_eland_query.txt
s_#lane_export.txt
This file contains the following content
[1. Machine]
[2. Run number]
3. Lane
4. Tile
5. X coordinate cluster
6. Y coordinate cluster
[7. index string]
[8. Read number (1 or 2 for paired-end reads, blank for single
read analysis)]
9. Read
10. Quality string. The ASCII character code = quality value +64
11. Match chromosome - name of the chromosome match or code
indicating why no match resulted.
12. Match contig - gives the contig name if there is a match and
the match chromosome is split into contigs (blank if no match is found)
13. Match position - always with respect to forward strand,
numbering starts at 1 (blank if no match found)
14. Match strand - "F" for forward, "R" for reverse
15. Match descriptor. Consice description of alignment. A
numerical denotes a run of matchine bases. A letter denotes
substitution of a nucleotide. Eg. 32C2 denotes substitution of a C at
the 33rd position.
16. Single read alignment score - alignment scor of a single-read
match, or for a paired-read, alignment scoire of a read if it were
treated as a single read.
[17. paired-read alignment score]
[18. partner chromosome - name of the chromosome if the read is
paired and its partner aligns to another chromosome]
[19. Partner Contig]
[20. Partner Offset]
[21. Partner Strand]
22. Filtering. Did the read pass quality filtering. Y for yes, N
for no.
An example fragment of such a file.
HWI-EAS264 303KWAAXX 1 1 638 200 GTNNNTTTTCTGCTTAGNNGTTTAATCATGTTTCAA NN???NNNNNNNNNNNN??NNNNNNNNNNNNNNNNN QC N
HWI-EAS264 303KWAAXX 1 1 1231 395 ACNNNCCAGAACGTGAANNAGCGTCCTGCGTGTAGC PN???NNNNNNNNNNNN??NNNNNNNNNNNNNNNNN QC N
HWI-EAS264 303KWAAXX 1 1 1061 436 GTNNNCCGCATGACCTTNNCCATCTTGGCTTCCTTT NN???NNNNNNNNNNNN??NNNNNNNNNNNNNNNNC QC N
HWI-EAS264 303KWAAXX 1 1 1076 412 GGNNNGTAGCGACAGCTNNGTTTTTAGTGAGTTGTT NN???NNNNNNNNNNNN??NNNNNNNNNNNNNNNNN QC N
HWI-EAS264 303KWAAXX 1 1 1785 543 GNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN N??????????????????????????????????? QC N
HWI-EAS264 303KWAAXX 1 1 552 176 GGNNNGTTATAACGCCGNNGCGGTAATAAACTCAAT NJ???NJNNNNNNJJNN??NNJNNNNBJNDDNBNEE QC N
HWI-EAS264 303KWAAXX 1 1 477 231 GTNNNGACAGCTTGGTTNNTAGTGAGTTTTTCCATT PN???NNNNNNNNNNNN??NNNNJNJNNDNNNNNNN QC N
HWI-EAS264 303KWAAXX 1 1 1598 633 GANNNTTTGACGGTTAANNGTGGTAATGGTGGTTTT PN???NNNNNNNNNNNN??HNNNNNNNNNNENENNN QC N
HWI-EAS264 303KWAAXX 1 1 653 186 GANNNTTTGCTATTCAGNNTTTGATGAATGCAATGC PN???NNNNNNNNNNNN??NNNNNNNNNNNNNNNEN QC N
HWI-EAS264 303KWAAXX 1 1 1066 418 GCNNNATGTTTACTCTTNNGCTTGTTCGTTTTCCGC NN???NNNNNNNNNNNN??NNNNJNNNNNNNNNNEN QC N
s_2_filt.txt
s_2_finished.txt
A file used by make to decide whether the process has finished or not.
s_2_frag.txt
s_2_percent_all.png
s_2_percent_base.png
s_2_percent_call.png
s_2_qcalreport.txt
s_2_qcal.txt
s_2_qraw.txt
s_2_qreport.txt
s_2_qtable.txt
s_2_saf.txt
s_2_score_files.txt
s_2_seqpre.txt
s_#lane_sequence.txt
Contains the sequences, quality scores and clusters after filtering. A
good strategy to filter out the sequences after filtering is grep
^[ACTG] <s_1_sequence.txt. The standard output is in fasq format
but with score+64 instead of score+32 to account for 'the dynamic
range', which doesn't make much sense anyway since a) nobody will
print these files; b) an extra 32 added will reduce the dynamic range
and since no scaling is reported this is probably nonsense.
@HWI-EAS264_303KWAAXX:4:1:978:308
GGTTGATATTTTTCATGGTATTGATAAAGCTGTTGC
+HWI-EAS264_303KWAAXX:4:1:978:308
]]]]]]]]]]]]]]]]][]]]][]]]][U][S[[P[
@HWI-EAS264_303KWAAXX:4:1:1246:245
GAAGTTAACACTTTCGGATATTTCTGATGAGTCGAA
+HWI-EAS264_303KWAAXX:4:1:1246:245
]]]]]]]]]]]]]]]][[]]]]][]U[][[[\\X\\
@HWI-EAS264_303KWAAXX:4:1:96:397
GTTCTGGTGATTCGTCTAAGAAGTTTAAGATTGCTG
+HWI-EAS264_303KWAAXX:4:1:96:397
]]]]]][]V]]]]V]]][[U[]V[]]]]R][[N[[M
s_2_Signal_Means.txt
s_2_sorted.txt
More Deep Sequencing notes
- http://analysis.yellowcouch.org/